MutAid Wiki
MutAid: Sanger and NGS based pipeline for mutation screening.
Status: Abandoned
Brought to you by:
ramvinay1
**The user manual of MutAid can be downloaded in PDF format from here...
**
MutAid supports three major NGS platforms including Illumina, Roche and Ion torrents.
MutAid supports Sanger sequencing data analysis from trace file to list of variants with extensive clinical annotation.
MutAid can be used to analyze the sequencing data generated from single-gene-panel, multigene-panel, exome-seq, and genome-seq experiments.
MutAid has uniform input and output model for Sanger and NGS data analysis.
MutAid supports five mappers including BWA, Bowtie, Bowtie2, TMAP and GSNAP to cover a wide range of NGS experiments.
MutAid supports four variant callers including GATK-HaplotypeCaller, Freebayes, SAMTOOLS and Varscan2 to identify the SNV and INDEL from NGS sequencing data.
MutAid can be used to analyze several patients/samples in a single run simultaneously.
To reduce the false positive and increase the sensitivity and specificity user can select the consensus variants from four variant callers and five mappers output.
Step 1 (Quality control and filtering) can be skipped if user has sequencing data in Sanger encoded FASTQ file format.
Pipeline can be started from Step 3 onwards if sequencing data is readily available in SAM/BAM format. This feature enables user to analyze NGS data produced from any sequencing platforms.
Linux
Mac OSX 10.6 or later
Python 2.7.9
Biopython 1.60 or higher
Perl 5.10 or higher
Java 1.7 or higher
R version 2.15.0 or higher
AlienTrimmer [ftp://ftp.pasteur.fr/pub/gensoft/projects/AlienTrimmer/]
TraceTuner [https://sourceforge.net/projects/tracetuner/]
*FASTQC [http://www.bioinformatics.babraham.ac.uk/projects/fastqc/]
*BWA [http://bio-bwa.sourceforge.net/]
Bowtie [http://bowtie-bio.sourceforge.net/index.shtml]
Bowtie2 [http://bowtie-bio.sourceforge.net/bowtie2/index.shtml]
TMAP [https://github.com/iontorrent/TS/tree/master/Analysis/TMAP]
GSNAP [http://research-pub.gene.com/gmap/]
GATK [https://www.broadinstitute.org/gatk/]
SAMTOOLS https://sourceforge.net/projects/samtools/
BCFTOOLS https://sourceforge.net/projects/samtools/files/samtools/1.2/bcftools-1.2.tar.bz2
Freebayes https://github.com/ekg/freebayes
Varscan2 http://varscan.sourceforge.net/
PICARD [http://broadinstitute.github.io/picard/]
*BedTools [https://github.com/arq5x/bedtools2]
genePredToGtf [http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/genePredToGtf]
Note: *denotes the Sanger analysis software/tools requirement
We provide source code in two version of MutAid for Linux and MAC-OSX computers. For Windows PC users we provide a fully configured Virtual Machine (VM can be used on any operating system). Along with source code and virtual machine we provide test data and extensive user manual for step-by-step get and run MutAid for expert and non-expert users.
For all users we provide a fully configured Virtual Machine (VM), which does not require any installation and configuration and works on any operating system including Windows, Linux and Mac osx. The VM can be obtained from https://sourceforge.net/p/mutaid/wiki/Virtual_Machine/
Step1.1: Download and install the Virtual Box from http://www.oracle.com/technetwork/server-storage/virtualbox/downloads/index.html#vbox
Step1.2: After installation of Virtual Box download and install the Virtual Box Extension Pack from http://www.oracle.com/technetwork/server-storage/virtualbox/downloads/index.html#extpack
Step2: Download MutAid Virtual machine from https://sourceforge.net/p/mutaid/wiki/Virtual_Machine
Step3: Import MutAid Virtual Machine file into Virtual Box.
Step4: Login into MutAid Virtual machine with username and password = testuser
Step5: Open terminal and change to MutAid_v1.0 directory with following command on terminal:
cd ~/MutAid_v1.0
Step6: Run MutAid pipeline with following command on terminal:
sh ./run_mutaid.sh
Step7: To run MutAid pipeline with other data set, prepare the Target file and MutAidOption file and run the MutAid
If user wants to use MutAid on own system then user can download the latest version of MutAid source code (1) for Linux computers from https://sourceforge.net/projects/mutaid/files/MutAid_v1.0-linux.zip and 2) for Macintosh OSX computers form https://sourceforge.net/projects/mutaid/files/MutAid_v1.0-macos.zip. Move the file to an appropriate directory and run the following command to uncompress the file:
unzip MutAid_v1.0-linux.zip
For Macintosh version run following command to unzip MutAid source code
unzip MutAid_v1.0-macos.zip
Note that after uncompressing the .zip file, a new folder will be created named MutAid_v1.0. This directory contains the following files and folders. Files are denoted in blue and sub folders are denoted in red colors:

To validate the installation of the MutAid pipeline, it can be run with a small test data set. The test data set and the corresponding MutAid configuration files for Sanger and NGS can be obtained from https://sourceforge.net/projects/mutaid/files/test_data.zip and download in MutAid_v1.0 folder/directory and run the following command to uncompress the file:
*unzip test_data.zip*
Note that after uncompressing the .zip file, a new folder will be created named test_data in MutAid_v1.0 folder. And then run following two commands to run Sanger data analysis and NGS data analysis.
Sanger analysis:
cd ~/MutAid_v1.0
./mutaid --option_file MutAidOptions_Sanger
NGS analysis:
cd ~/MutAid_v1.0
./mutaid --option_file MutAidOptions_NGS
To get help on how to run MutAid and required parameters enter:
./mutaid –help
MutAid pipeline consists of six sequential steps, which can be run by a single command. All input parameters can be specified in the MutAidOptions file. These parameters are then used to run the whole pipeline. MutAid provides two different input options file for Sanger and NGS:
MutAidOptions_Sanger: This configuration file can be used for Sanger sequencing data analysis for mutation screening. We have already given default value for required parameters to run the whole Sanger sequencing analysis from raw reads to variation list.
MutAidOptions_NGS: This configuration file can be used for NGS sequencing data analysis for mutation screening. We have already given default value for required parameters to run the whole NGS sequencing analysis from raw reads to variation list.
MutAid can be run with the following command, which should be run under the folder/directory MutAid_v1.0 directory:
./mutaid --option-file <path to MutAidOptions file>
However, before running MutAid with your own dataset some parameters need to be changed in the appropriate MutAid Options files (MutAidOptions_Sanger OR MutAidOptions_NGS).
Before starting the data analysis with MutAid, user need to prepare reference information files (genome FASTA sequence, gene annotation, and variant information) by using the “prepref” tool, which is available within MutAid pipeline. prepref downloads RefSeq gene annotation and linked database cross-reference ID of various databases from the UCSC Table browser (http://genome.ucsc.edu/cgi-bin/hgTables). The reference files need to be prepared only once.
Thus following three steps are required to use MutAid with your Sanger or NGS data analysis.
MutAid uses RefSeq reference genome FASTA file, RefSeq gene annotation file and other SNP and INDEL annotation files from UCSC table browser. We provide a tool in MutAid to do it automatically by running the following commands.
Go to MutAid source directoy with following command:
cd ~/MutAid_v1.0
Run following command to prepare reference files by giving two parameters:
(1) Genome assembly hg19 or hg38
(2) dbSNP build number 137 or 141 or 142
./prepref --genome_assembly hg19 --dbSNP_version 142
After successful completion of this tool it creates a ref_input folder in MutAid_v1.0 folder. The path of reference folder is MutAid_v1.0/ref_input
In this step following reference files will be downloaded and prepared for MutAid analysis. Note that this command needs to be run only in the beginning. Afterwards MutAid can use these files for all Sanger and NGS data analysis.
1. Genome FASTA file
2. RefSeq Genome Gene annotation GTF
3. Genome Gene database cross references
4. Genome SNP and INDEL
5. GATK Bundle (only used for NGS data analysis)
Step1: Prepare Input files:
1. Prepare Target file
For each analysis user need to prepare a target file in a predefined format. It is a tab-separated text file, which contains 10 columns. As shown in figure 1, one row for each sequencing file in target file. Target file is a mandatory input, which must be provided. The target file can be given in the MutAidOptions file with the input name Target_File=”sanger_target_file.txt”
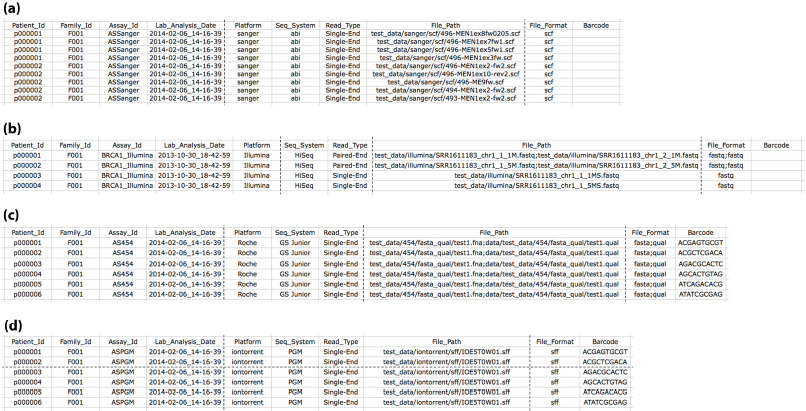
Figure 1: Shows Target files (a) Sanger target file, (b) Illumina target file, (c) 454 target file, (d) Ion torrent target
2. Prepare MutAidOptions_Sanger file
MutAid requires an input configuration file, which can be prepared once by customizing parameters as per the requirement, and the whole pipeline will be run without further user interaction. The MutAid Option file is shown below:
##################################################################################################################################
## MutAid Options file for Sanger data analysis (MutAidOptions_Sanger)
##
## Note: This configuration file is used for Sanger data analysis.
##
## This file sets the customizable options for running MutAid-Sanger data analysis. The options include the paths to the
## database cross reference files, paths to executables, annotation files and other parameters of the pipeline
##
## This file contains example parameter settings, which can be used to run the test data set
## provided with MutAid.
## For running MutAid with your own data, the example parameter values have to be modified.
## Please note that only the input value but not the parameter name itself should be changed.
##
## Note: 1) Lines starting with # are comment lines. They do not need to be changed.
## 2) Parameter's name is case sensitive. The parameters can be changed
## by replacing the values after the “=” sign.
##
##################################################################################################################################
##################################################################################################################################
#
### Global input parameters
#
##################################################################################################################################
# Give the path to the fasta file of the genomic reference. The reference fasta should be obtained from UCSC genome browser
REFERENCEE_FASTA = "ref_input/hg19_fasta/hg19.fa"
# Give the gene annotation file of your genomic reference in GTF format. The reference GTF should be obtained from UCSC genome browser
REFERENCE_GTF_FILE = "ref_input/hg19_annotation/hg19_refseqgenes.gtf"
### Give the number of processors that should be used in MutAid pipeline.
THREAD = 2
### Reference genome information from UCSC genome browser
Genome_Assembly = hg19
Refseq_Genome_Build = GRCh37
### dbSNP version from UCSC genome browser
dbSNP_Version = 142
##################################################################################################################################
#
# Provide the experiment information files
#
##################################################################################################################################
### Target_File contains Sequencing information. (mandetory). CAN BE EDITED
Target_File = "test_data/sanger/scf/test_target_file.txt"
### For targeted resequencing or Sanger sequencing provide the assay information file in GTF format (optional). CAN BE EDITED
Assay_Gtf_File = ""
### A comma seperated list of chromosomes, in case dont need to use full genome FASTA sequences an GTF annotation. CAN BE EDITED
### This is optional and is extremely useful for targeted sequencing.
Target_Chromosome = chr11
### Provide the Hotspot mutation in bed format which will be co-analyzed. (optional). CAN BE EDITED
Hotspot_Bed_File = ""
### Provide the Primer and adapter sequences for trimming. (optional). CAN BE EDITED
Primer_Adapter_File = ""
##################################################################################################################################
#
# Output file and directory location
#
##################################################################################################################################
### Provide the output directory. CAN BE EDITED
OUTPUT_DIRECTORY = "test_output/sanger_output_dir"
### Provide the final output file name. CAN BE EDITED
#OUTPUT_FILE = "test_output/sanger_output_file"
##################################################################################################################################
#
### Step1: Quality control and filtering
#
##################################################################################################################################
### Minimum average base quality for trimming the low quality 5' and 3' end of reads
Minimum_Base_Quality = 20
### Minimum read length to discard the read for further analysis
Minimum_Read_Length = 50
### Maximum read length to discard the read for further analysis
Maximum_Read_Length = 1000
### Sanger data quality control and trimming parameters for TTUNER
Sanger_Trim_Window_Size = 10
Sanger_Trim_Base_Quality = 20
##################################################################################################################################
#
### Step2: Mapping to reference genome
#
##################################################################################################################################
### Mapper name only one mapper at a time allowed. Example Mapper_Name = bwa
Mapper_Name = bwa
### Provide the Mapper_Parameters
Mapper_Parameters = ""
BWA_Sampe_Parameters = ""
### Filtering Mapped reads from BAM/SAM file
Minimum_Mapping_Quality = 20
##################################################################################################################################
#
### Step3: Variant detection
#
##################################################################################################################################
### Variant callers name.
Variant_Caller = samtools
### SNV and INDEL calling parameters
### Minimum read coverage to call variant
Minimum_Coverage = 4
### Maximum read coverage to call variant. This is useful to discard the repetative region.
Maximum_Coverage = 1000
### Minimum Variant allele read count to consider as variant position
Minimum_Count = 2
### Minimum allele frequency of Variant allele to consider as variant position
Minor_Allele_Frequency = 0.01
### Provide the Hotspot mutation in bed format which will be co-analyzed. (optional)
Hotspot_Bed_File = ""
##################################################################################################################################
#
### Step4: Variant effect prediction
#
##################################################################################################################################
##################################################################################################################################
#
### Step5: Variant functional annotation
#
##################################################################################################################################
## Various SNPs information file from UCSC Table browser.
## Provide the full path to all these files.
UCSC_All_SNPs = ref_input/hg19_annotation/hg19_snpAll.txt
UCSC_Common_SNPs = ref_input/hg19_annotation/hg19_snpCommon.txt
UCSC_Flagged_SNPs = ref_input/hg19_annotation/hg19_FlaggedSNPs.txt
UCSC_HapMap_SNPs = ref_input/hg19_annotation/hg19_HapMapSNPs.txt
UCSC_Multi_SNPs = ref_input/hg19_annotation/hg19_MultSNPs.txt
UCSC_COSMIC_SNPs = ref_input/hg19_annotation/hg19_cosmic.txt
UCSC_GWAS_Catalog_SNPs = ref_input/hg19_annotation/hg19_gwasCatalog.txt
UCSC_CpG_Islands_SNPs = ref_input/hg19_annotation/hg19_cpgIslandExt.txt
UCSC_Coding_DbSnp = ref_input/hg19_annotation/hg19_CodingDbSnp.txt
UCSC_OrthoPt3Pa2Rm2_SNPs = ref_input/hg19_annotation/hg19_OrthoPt3Pa2Rm2.txt
UCSC_RefseqGene_Info = ref_input/hg19_annotation/hg19_refseqgenes_dbxref.gtf
##################################################################################################################################
#
### Step6: Producing variant summary output
#
##################################################################################################################################
### Give the number of based to show in flanking region for each variant in UCSC browser and Ensemble browser
Flanking_Region = 100
##################################################################################################################################
#
# Third party software/tool executables
# Provide the full path to the third party executables to run the MutAid pipeline.
#
#
##################################################################################################################################
### Get samtools to manipulate SAM and BAM files from http://samtools.sourceforge.net/
# Provide the full path of the executables relative to the <MutAid_1.0>
###### SAMTOOLS path
SAMTOOLS = executables/samtools-0.1.19/samtools
BCFTOOLS = executables/samtools-0.1.19/bcftools/bcftools
### Define the full path of the Mappers
BWA = executables/bwa-0.7.9a/bwa
###### FASTQC executables
FASTQC = executables/FastQC/fastqc
###### Sanger ab1 and scf file QC tool
TTUNER = executables/tracetuner_3.0.6beta/rel/Linux_64/ttuner
AlienTrimmer = executables/AlienTrimmer_0.4.0/src/AlienTrimmer.jar
#### PICARD JAR file from http://picard.sourceforge.net/index.shtml
###### 3 PICARD jar files are required for MutAid pipeline
PICARD = executables/picard-tools-1.130/picard.jar
### Get intersectBed executables from a collection of useful utilities called bedtools-
### from http://code.google.com/p/bedtools/
###### 3 bedtools
intersectBed = executables/bedtools2/bin/intersectBed
bedtools = executables/bedtools2/bin/bedtools
bamToBed = executables/bedtools2/bin/bamToBed
Step2: Run MutAid pipeline:
After preparing the Target File and MutAidOptions file have been prepared and customized then MutAid pipeline can be run with following command line
./mutaid --option_file MutAidOptions_Sanger
Step1: Prepare Input files:
1. Prepare Target file
For each analysis, user need to prepare a target file in a predefined format. It is a tab-separated text file, which contains 10 columns. As shown in Figure 1a-c above, one row for each sequencing file in target files. Target file is a mandatory input, which must be provided. The target file can be given in the MutAidOptions file with the input name Target_File=”ngs_target_file.txt”
2. Prepare MutAidOptions_NGS file
MutAid requires an input configuration file, which can be prepared once by customizing parameters as per the requirement, and the whole pipeline will be run without further user interaction. The MutAid Option file is shown below:
##################################################################################################################################
## MutAid Options file for NGS data analysis (MutAidOptions_NGS)
##
## Note: This configuration file is used for NGS data analysis.
##
## This file sets the customizable options for running MutAid-NGS data analysis. The options include the paths to the
## database cross reference files, paths to executables, annotation files and other parameters of the pipeline
##
## This file contains example parameter settings, which can be used to run the test data set
## provided with MutAid.
## For running MutAid with your own data, the example parameter values have to be modified.
## Please note that only the input value but not the parameter name itself should be changed.
##
## Note: 1) Lines starting with # are comment lines. They do not need to be changed.
## 2) Parameter's name is case sensitive. The parameters can be changed
## by replacing the values after the “=” sign.
##
##################################################################################################################################
##################################################################################################################################
#
### Global input parameters
#
##################################################################################################################################
# Give the path to the fasta file of the genomic reference. The reference fasta should be obtained from UCSC genome browser.
REFERENCEE_FASTA = "ref_input/hg19_fasta/hg19.fa"
# Give the gene annotation file of your genomic reference in GTF format. The reference GTF should be obtained from UCSC genome browser.
REFERENCE_GTF_FILE = "ref_input/hg19_annotation/hg19_refseqgenes.gtf"
### Give the number of processors that should be used in MutAid pipeline. CAN BE EDITED
THREAD = 4
### Reference genome information from UCSC genome browser
Genome_Assembly = hg19
Refseq_Genome_Build = GRCh37
### dbSNP version from UCSC genome browser
dbSNP_Version = 142
##################################################################################################################################
#
# Provide the experiment information files
#
##################################################################################################################################
### Target_File contains Sequencing information. (mandetory). CAN BE EDITED
Target_File = "test_data/illumina/Illumina_target_file.txt"
#Target_File = "test_data/illumina/BAM_target_file.txt"
### For targeted resequencing or Sanger sequencing provide the assay information file in GTF format (optional) CAN BE EDITED
Assay_Gtf_File = ""
### A comma seperated list of chromosomes, in case dont need to use full genome FASTA sequences an GTF annotation. CAN BE EDITED
### This is optional and is extremely useful for targeted sequencing.
Target_Chromosome = chr13,chr17
### Provide the Hotspot mutation in bed format which will be co-analyzed. (optional) CAN BE EDITED
Hotspot_Bed_File = ""
### Provide the Primer and adapter sequences for trimming. (optional) CAN BE EDITED
Primer_Adapter_File = ""
##################################################################################################################################
#
# Output file and directory location
#
##################################################################################################################################
### Provide the output directory. CAN BE EDITED
OUTPUT_DIRECTORY = "test_output/ngs_output_dir"
### Provide the final output file name. CAN BE EDITED
#OUTPUT_FILE = "test_output/454_output_file"
##################################################################################################################################
#
### Step1: Quality control and filtering
#
##################################################################################################################################
### Minimum average base quality for trimming the low quality 5' and 3' end of reads
Minimum_Base_Quality = 20
### Minimum read length to discard the read for further analysis
Minimum_Read_Length = 50
### Maximum read length to discard the read for further analysis
Maximum_Read_Length = 1000
##################################################################################################################################
#
### Step2: Mapping to reference genome
#
##################################################################################################################################
### Mapper name only one mapper at a time allowed. Example Mapper_Name = bwa. CAN BE EDITED
Mapper_Name = bwa,bowtie2
### Provide the Mapper_Parameters it could be any mapper's parameter
Mapper_Parameters = ""
BWA_Sampe_Parameters = ""
### Filtering Mapped reads from BAM/SAM file
Minimum_Mapping_Quality = 20
##################################################################################################################################
#
### Step3: Variant detection
#
##################################################################################################################################
### Variant callers name. The default one is samtools. CAN BE EDITED
Variant_Caller = varscan,samtools
### SNV and INDEL calling parameters
### Minimum read coverage to call variant
#Minimum_Coverage = 20
Minimum_Coverage = 20
### Maximum read coverage to call variant. This is useful to discard the repetative region.
Maximum_Coverage = 1000
### Minimum Variant allele read count to consider as variant position
#Minimum_Count = 4
Minimum_Count = 4
### Minimum allele frequency of Variant allele to consider as variant position
Minor_Allele_Frequency = 0.01
### Provide the Hotspot mutation in bed format which will be co-analyzed. (optional)
Hotspot_Bed_File = ""
##################################################################################################################################
#
### Step4: Variant effect prediction
#
##################################################################################################################################
##################################################################################################################################
#
### Step5: Variant functional annotation
#
##################################################################################################################################
## Various SNPs information file from UCSC Table browser
## Provide the full path to all these files.
UCSC_All_SNPs = ref_input/hg19_annotation/hg19_snpAll.txt
UCSC_Common_SNPs = ref_input/hg19_annotation/hg19_snpCommon.txt
UCSC_Flagged_SNPs = ref_input/hg19_annotation/hg19_FlaggedSNPs.txt
UCSC_HapMap_SNPs = ref_input/hg19_annotation/hg19_HapMapSNPs.txt
UCSC_Multi_SNPs = ref_input/hg19_annotation/hg19_MultSNPs.txt
UCSC_COSMIC_SNPs = ref_input/hg19_annotation/hg19_cosmic.txt
UCSC_GWAS_Catalog_SNPs = ref_input/hg19_annotation/hg19_gwasCatalog.txt
UCSC_CpG_Islands_SNPs = ref_input/hg19_annotation/hg19_cpgIslandExt.txt
UCSC_Coding_DbSnp = ref_input/hg19_annotation/hg19_CodingDbSnp.txt
UCSC_OrthoPt3Pa2Rm2_SNPs = ref_input/hg19_annotation/hg19_OrthoPt3Pa2Rm2.txt
UCSC_RefseqGene_Info = ref_input/hg19_annotation/hg19_refseqgenes_dbxref.gtf
##################################################################################################################################
#
### Step6: Producing variant summary output
#
##################################################################################################################################
### Give the number of based to show in flanking region for each variant in UCSC browser and Ensemble browser
Flanking_Region = 100
##################################################################################################################################
#
# Third party software/tool executables
# Provide the full path to the third party executables to run the MutAid pipeline.
#
#
##################################################################################################################################
### Get samtools to manipulate SAM and BAM files from http://samtools.sourceforge.net/
# Provide the full path of the executables relative to the <MutAid_1.0>
###### SAMTOOLS path
SAMTOOLS = executables/samtools-0.1.19/samtools
BCFTOOLS = executables/samtools-0.1.19/bcftools/bcftools
### Define the full path of the Mappers
BWA = executables/bwa-0.7.9a/bwa
TMAP = executables/TMAP/tmap
GSNAP = executables/gmap-2014-12-28/bin/gsnap
BOWTIE = executables/bowtie-1.1.2/bowtie
BOWTIE2 = executables/bowtie2-2.2.5/bowtie2
###### FASTQC executables
FASTQC = executables/FastQC/fastqc
###### AlienTrimmer for Adapter and Primer and Homopolymer trimming
AlienTrimmer = executables/AlienTrimmer_0.4.0/src/AlienTrimmer.jar
###### varient callers path
#GATK = executables/GenomeAnalysisTK.jar
GATK_BUNDLE = ref_input/hg19_gatk_bundle
FreeBayes = executables/freebayes/bin/freebayes
Varscan2 = executables/VarScan.v2.3.7.jar
#### PICARD JAR file from http://picard.sourceforge.net/index.shtml
###### 3 PICARD jar files are required for MutAid pipeline
PICARD = executables/picard-tools-1.130/picard.jar
### Get intersectBed executables from a collection of useful utilities called bedtools-
### from http://code.google.com/p/bedtools/
###### 3 bedtools
intersectBed = executables/bedtools2/bin/intersectBed
bedtools = executables/bedtools2/bin/bedtools
bamToBed = executables/bedtools2/bin/bamToBed
### Filter the variants by minimum number of supported mappers. CAN BE EDITED
Minimum_Number_of_supported_mapper=2
### Filter the variants by minimum number of supported variant callers. CAN BE EDITED
Minimum_Number_of_supported_vc=2
Step2: Run MutAid whole pipeline:
After preparing the Target File and MutAidOptions file have been prepared and customized then MutAid pipeline can be run with following command line
./mutaid --option_file MutAidOptions_NGS
Step2: Run MutAid pipeline step-by-step:
Alternatively user can run MutAid pipeline form NGS data analysis in a step-by-step manner. The main advantage of this feature is, user can optimize the parameters for each step independently without spending time to finish whole pipeline. Once all parameters have been optimized then providing the MutAidOptions_NGS file can run whole pipeline.
Since NGS data are enormous in size and in coverage thus we have facilitated MutAid whole pipeline with many start and stop points and thus user can run 1) Only quality control and filtering step, 2) Only Mapping step and 3) Only variant calling and 4) Only Variant effect prediction, variant annotation and write final variant summary table output.
Run MutAid only for Quality control and filtering
./mutaid --option_file MutAidOptions_NGS --qc
Run MutAid only for Mapping
./mutaid --option_file MutAidOptions_NGS --map
Run MutAid only variant detection
./mutaid --option_file MutAidOptions_NGS --variant_call
Run MutAid for only writing output
With this command MutAid 1) predicts genomic effects like codon change, amino acid change and genomic feature assignment 2) Functional and clinical annotation of all resulting variants and 3) write final output variant summary table.
./mutaid --option_file MutAidOptions_NGS --write_output
After running MutAid, the results of the MutAid pipeline can be found in the output directory (as specified in the MutAid Options file). Results are provided in the following format.
<OUTPUT_FILE>
<OUTPUT_DIR>
|
- <fastq_files_after_qc>
|
- <patient1_1.fq;patient1_2.fq>
- <patient2_1.fq;patient2_2.fq>
- <patient3_1.fq;patient3_2.fq>
- .
- .
- .
- <patientN_1.fq;patientN_2.fq>
- <QC_report>
|
- <before_qc_patient1_1.fq_fastqc_qc_report.html>
- <after_qc_patient1_1.fq_fastqc_qc_report.html>
- <before_qc_patient1_2.fq_fastqc_qc_report.html>
- <after_qc_patient1_2.fq_fastqc_qc_report.html>
- .
- .
- . |
- .
- <before_qc_patientN_1.fq_fastqc_qc_report.html>
- <after_qc_patientN_1.fq_fastqc_qc_report.html>
- <before_qc_patientN_2.fq_fastqc_qc_report.html>
- <after_qc_patientN_2.fq_fastqc_qc_report.html>
- < bam_files>
|
- <patient1.bam>
- <patient2.bam>
- <patient3.bam>
- .
- .
- .
- <patientN.bam>
- <variant_files>
|
- <patient1.vcf>
- <patient2.vcf>
- <patient3.vcf>
- .
- .
- .
- <patientN.vcf>
<fastq_files_after_qc>:</fastq_files_after_qc>
This output folder contains high quality FASTQ file for each patient with Sanger quality encoding. If reads are in paired-end then there will be two files for each patient.
<qc_report>:</qc_report>
This output folder contains Quality control and trimming report in html format generated by FASTQC tool. There are two files for each FASTQ files 1) before quality control and 2) after quality control.
<bam_files>:</bam_files>
This output folder contains resulting BAM file along with BAM index for each patient/sample. These BAM files have been generated after applying all mapping parameters and post-mapping filtering criteria.
<variant_files>:</variant_files>
This output folder contains resulting variants (SNV, Insertion, and Deletion) in Variant Call Format (VCF) for each patient/sample.
Ram Vinay Pandey
ramvinay.pandey@gmail.com
