DaMold Wiki
a data mining platform for variant annotation and visualization.
Brought to you by:
ramvinay1
DaMold a powerful, integrated, web-based, and user-friendly tool to filter, annotate, narrow down variant for target genes or transcripts, cross-link, and visualize NGS, Sanger, and hotspot variants.
DaMold allows to narrow down the analysis to list of genes, list of transcripts, variant of only coding region, variant with only amino acid change or variants with only splicing effects.
It is an easy-to-use software, which provides flexible input options and accepts variants in VCF (version 4.1 and 4.2) and standard BED formats.
For each variant DaMold predicts the variant effect, such as codon change, and amino acid change.
Next, it cross-links each variant with 36 clinically relevant public databases, which contain already reported SNPs and INDELs from previous experimental studies along with associated genomic, proteomic, and clinical information.
DaMold, seamlessly integrates four widely used genome browser such as the UCSC genome browser , Ensembl genome browser, 1000 Genomes browser, and NCBI variation viewer.
Each variant is directly linked with each genome browser.
DaMold can be used to analyze, elucidate, and interpret variants from data generated by targeted re-sequencing, gene-panel sequencing, exome, and whole genome sequencing.
DaMold is a user-friendly and web-based tool, which is available online as web app and as Docker container which can be easily run on local server.
Operating system
1. Linux Ubuntu 14.04 or higher (64bit)
2. Mac OS X 10.10.3 Yosemite or newer (64bit)
3. Windows 10 Pro, or newer (64bit)
Softwares requirments
1. Docker version 1.12.0 (https://www.docker.com/)
2. Python 2.7.12 or higher (https://www.python.org/)
3. Biopython 1.67 or higher (http://biopython.org/)
4. Perl 5.18.2 or higher (https://www.perl.org/)
5. Apache (version 2.4.7) webserver (https://httpd.apache.org/)
6. Virtual Box (version 5.1.2 or higher) (http://www.oracle.com/technetwork/server-storage/virtualbox/downloads/index.html#vbox)
7.
Tools used by DaMold
1. BedTools (version 2.26.0) (https://github.com/arq5x/bedtools2)
2. SamTools (version 1.3.1) (https://sourceforge.net/projects/samtools)
3. UCSC tool genePredToGtf (http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/genePredToGtf)
4. SnpEff (version 4.2) (http://snpeff.sourceforge.net/)
Note: No manual installation of above tools are required.
The oneline Web version of DaMold can be run via http://damold.platomics.com/index.html
To run DaMold on local server, source code is available as Docker container. The DaMold Docker is available here.
DaMold is also available as fully configured VM and can be run on local computer. The DaMold virtual machine is available here.
User can filter, annotate, cross-reference and visualize raw variants with DaMold by providing input options and files from query page as shown in Figure 1.
Following input parameters and input files are required.
Input 1:
Genome information parameters are required to download genome fasta sequence and annotation files from UCSC genome browser (http://genome.ucsc.edu/index.html)
1) Select genome name from first drop down menu (human or mouse). The default genome is human.
2) Select genome assembly version from second drop down menu (default genome assembly is hg19)
3) Select the dbSNP build to download the corresponding common SNPs from UCSC genome browser. Default is 142 for human, genome assembly hg19.
Input 2:
Upload an input variant file in VCF (version 4.1 and 4.2) or BED format. This is mandetory input file.
Input 3:
Hotspot mutation input file in VCF (version 4.1 and 4.2) or BED format. This is optional input.
Input 4:
To narrow down variants of region of interest (for list of genes, list of transcripts), only coding region variants, only variants with amino acid changes or variants only with splicing effects.
Input 5:
To filter the low confidence variants, DaMold provides various useful variant selection parameters. This is optional to filter the variants. By default DaMold does not filter the variants. To filter variants user need to check the checkbox beside the Parameters for variant selection Under these parameters user can define:
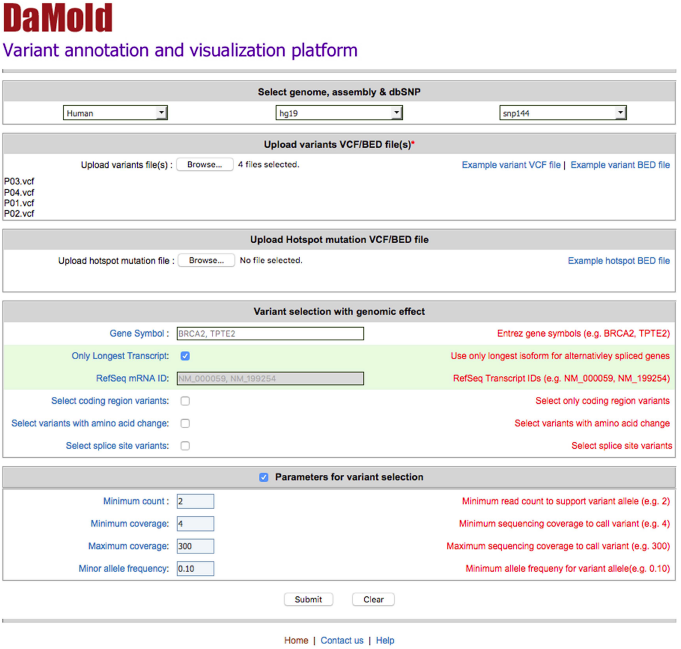
Figure 1 - Web interface of DaMold query page.
The query interface of DaMold shows different parameters that can be used to download corresponding reference genome annotations and variant annotations. Variants are accepted in VCF or BED format. Hotspot mutations can be uploaded in BED format.
The output web interface of DaMold provides a user-friendly output summary table, which links all identified variants with several databases. Instead of opening a new browser window and redirecting the user to the corresponding page, DaMold conveniently displays the information from the linked resource in a panel below the output table.
An example output page is shown in Figure 2 below, where the whole output page is divided into 3 panels. The upper panel shows the loaded variant summary table along with the direct hyperlinks to the linked databases. The middle panel is for navigating the summary table, where 20 variants can be displayed at a time. The bottom panel loads all integrated database pages in the same window, so that users can visualize the variant summary table and the linked pages simultaneously. On top of the page, DaMold provides links to download the complete variant table in TXT and HTML format.
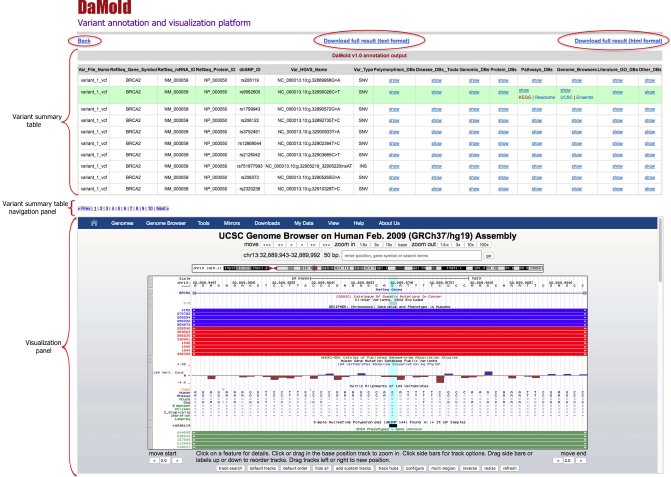
Figure 2 - DaMold variant summary output table
DaMold produces a final variant summary output in HTML format, where each line contains one variant including variant information, variant effects and cross-references to external databases. The output page displays three panels: the top panel is the variant summary table which shows 20 variants at a time; the middle panel is the variant table navigation panel; the bottom panel is used to load and view information from linked databases.
DaMold cross-links previously described polymorphism, disease, genetic, protein, gene function, and pathway information, which could help to select the clinically relevant variants. As shown below, DaMold cross-references each variant with 33 relevant publically available databases and genome browsers (see Figure 3). The resources integrated in the DaMold graphical user interface are described below:
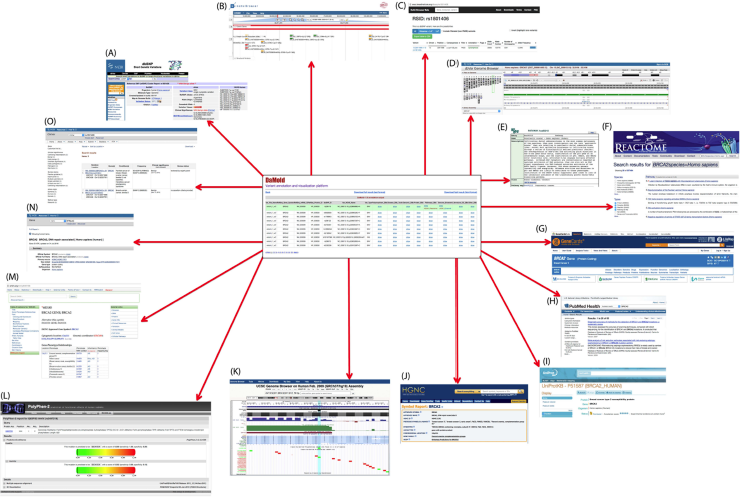
Figure 3 – Cross-referenced databases
(A) PolyPhen-2, (B) KEGG pathway, (C) Ensembl genome browser, (D) UCSC genome browser, (E) Uniprot, (F) Pubmed, (G) Reactome pathway, (H) Entrez gene, (I) GeneCards, (J) COSMIC and (K) dbSNP.
DaMold provides extensive annotation and cross-references for each variant with 33 clinically relevant databases.
DaMold provides previously described polymorphism, disease, genetic, protein, gene function, and pathway information, which could help to select the clinically relevant variants. As listed below all databases, DaMold integrates all these databases under single interface (see Figure 3).
Polymorphism databases
dbSNP: http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs=rs1800354
dbVar: http://www.ncbi.nlm.nih.gov/dbvar/browse/org/?taxid=9606&assm=GCF_000001405.13&chr=13&from=23898663&to=23898664
NCBI Variation Viewer: http://www.ncbi.nlm.nih.gov/variation/view/?chr=13&from=23898663&to=23898664&assm=GCF_000001405.13
1000 Genomes: http://browser.1000genomes.org/Homo_sapiens/Location/View?r=13:23898663-23898664
SNPedia: http://www.snpedia.com/index.php/rs1800354
ExAC: http://exac.broadinstitute.org/dbsnp/rs1800354
Disease databases and tools
ClinVar: http://www.ncbi.nlm.nih.gov/clinvar?term=rs1800354
COSMIC: http://grch37-cancer.sanger.ac.uk/jbrowse/index.html?tracks=cosmic_genes,mutations,breakpoints&loc=13:23898663..23898664&data=data/json/grch37/v77/cosmic
NCBI GTR: http://www.ncbi.nlm.nih.gov/gtr/genes/6445
OMIM: http://omim.org/entry/608896
DECIPHER: https://decipher.sanger.ac.uk/browser#q/13:23898663-23898664/location/13:23898663-23898664
GWAS CENTRAL: http://www.gwascentral.org/search?q=chr13:23898663..23898664
PolyPhen-2:http://genetics.bwh.harvard.edu/cgi-bin/pph2/dbsearch.cgi?varspec=rs1800354
Genomic databases
Entrez Gene: http://www.ncbi.nlm.nih.gov/gene/?term=6445
RefSeq mRNA: http://www.ncbi.nlm.nih.gov/nuccore/NM_000231
HomoloGene: http://www.ncbi.nlm.nih.gov/homologene/?term=NM_000231
GEO Profiles: http://www.ncbi.nlm.nih.gov/geoprofiles/?term=NM_000231
UniGene: http://www.ncbi.nlm.nih.gov/unigene/?term=NM_000231
HGNC: http://www.genenames.org/data/hgnc_data.php?hgnc_id=10809
Protein databases
Refseq_Protein: http://www.ncbi.nlm.nih.gov/protein/NP_000222
UniProt: http://www.uniprot.org/uniprot/Q13326
Pathways databases
KEGG: http://www.genome.jp/dbget-bin/www_bget?pathway:hsa05416
Reactome: http://www.reactome.org/content/query?q=SGCGspecies=Homo+sapiens&types=Complex&types=Protein&types=Pathway&types=Reaction&cluster=true
Genome browsers
UCSC: http://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&position=chr13:23898663-23898664&pix=1200
Ensembl: http://grch37.ensembl.org/Homo_sapiens/Location/View?db=core;r=13:23898663-23898664
Literature & GO databases
Pubmed: http://www.ncbi.nlm.nih.gov/pmc/?term=NM_000231
Pubmed Health: http://www.ncbi.nlm.nih.gov/pubmedhealth/?term=SGCG
MedGen: http://www.ncbi.nlm.nih.gov/medgen/?term=SGCG
GOPubmed: http://www.gopubmed.org/web/gopubmed/search?t=hgnc&q=SGCG
QuickGO: http://www.ebi.ac.uk/QuickGO/GProtein?ac=Q13326
Other databases
WikiGenes: http://www.wikigenes.org/e/gene/e/6445.html
GeneTes: https://www.genetests.org/genes/?gene=SGCG
BioGPS: http://biogps.org/#goto=genereport&id=6445
GENATLAS: http://genatlas.medecine.univ-paris5.fr/fiche.php?symbol=SGCG
GeneCards: http://www.genecards.org/cgi-bin/carddisp.pl?gene=SGCG
H_InvDB: http://biodb.jp/hfs.cgi?db1=HUGO&type=GENE_SYMBOL&db2=Locusview&id=SGCG
To validate the proper function of the DaMold tool it can be run with a small test data set. The test data set can be obtained from https://sourceforge.net/projects/damold/files/test_data.zip
and run the following command to uncompress the file:
unzip test_data.zip
Note that after uncompressing the .zip file, a new folder will be created named test_data. Now upload these files on the DaMold query page (http://localhost/damold) and run the primer design.
DaMold requires input variants in VCF or BED format as described below:
DaMold accepts input variant which are obtained from NGS and Sanger sequencing data analysis in VCF (http://www.1000genomes.org/wiki/Analysis/vcf4.0/) format (version 4.1 and 4.2). It must be valid VCF file. An example VCF file is given in test data set https://sourceforge.net/projects/damold/files/test_data.zip
DaMold also accepts input in BED (http://www.ensembl.org/info/website/upload/bed.html) format. The BED format input consists of three mandatory columns (Chromosome, Chromosome start and Chromosome end) and two optional columns (Reference allele and Variant allele). In example data shown below:
Column 1) chromosome, Column 2) chromosome start position, Column 3) chromosome end position, 4) reference allele, and 5) variant allele. First 3 columns are mandetory.
chr13 32889967 32889968 G A
chr13 32890025 32890026 C T
chr13 32890571 32890572 G A
chr13 32892734 32892735 T C
chr13 32900932 32900933 T A
chr13 32902393 32902394 T C
chr13 32903684 32903685 C T
chr13 32905218 32905219 A AT
chr13 32905264 32905265 G A
chr13 32910327 32910328 T C
chr13 32911887 32911888 A G
chr13 32913054 32913055 A G
chr13 32915004 32915005 G C
chr13 32915409 32915410 CAATT C
chr13 32915523 32915524 C A
chr13 32918940 32918941 A G
chr13 32918966 32918967 G A
chr13 32920843 32920844 T C
chr13 32928316 32928317 T C
chr13 32928403 32928404 T C
chr13 32928407 32928408 G A
chr13 32929231 32929232 A G
chr13 32929386 32929387 T C
chr13 32930935 32930936 G A
DaMold can co-analyze the known hotspot variants along with raw variants obtained from NGS and Sanger data analysis. Hotspot mutation can be provided in VCF or BED format an example BED format is shown below:
chr13 20000129 20000130 G T chr13 20000190 20000191 C T chr13 20000342 20000343 G T chr13 20000416 20000417 C T chr13 20000629 20000630 T C chr13 20000701 20000702 G C chr13 32889967 32889968 G A chr13 32890025 32890026 C T chr13 32890571 32890572 G A chr13 32892734 32892735 T C chr13 32900932 32900933 T A chr13 32963706 32963707 C T chr13 32965650 32965651 T C chr13 32968590 32968591 G A chr13 32968606 32968607 A G
False positive and false negative rates are vital for clinical NGS applications, and are significantly dependent on the level of sequencing coverage. Thus, DaMold filters the raw variants based on minimum coverage of variant allele, minimum total coverage, maximum total coverage (to avoid repetitive or duplicated regions), and minimum variant allele frequency.
DaMold provide different parameters to filter the low confidence variants from further downstream analysis can be set. The default filtering parameters on the query page are:
Minimum coverage of variant allele: 2
Minimum total coverage: 4
Maximum total coverage: 300
Minimum minor allele frequency: 0.10.
To use DaMold on local computer for variant annotation, obtain the virtual machine, open web browser and type the following url into browser (http://localhost/damold) and upload the variant file in VCF or BED format. To filter varitns provide appropriate cutoff for given parameters.
To evaluate and demonstrate the performance of the DaMold pipeline for variant annotation and visualization of variants generated from the analysis of NGS data. We used publicly available variant VCF files produced from Human Whole Exome Sequencing (WES) of a CEPH/UTAH female individual (HapMap ID: NA12878) was sequenced under the Genome in a Bottle Consortium (NIST) project. We downloaded VCF file from ftp://ftp-trace.ncbi.nih.gov/giab/ftp/data/NA12878/analysis/Illumina_PlatinumGenomes_NA12877_NA12878_09162015/hg19/8.0.1/NA12878/NA12878.vcf.gz.
We randomly sampled variants ranging from 10 to 200,000, which could be analyzed in ~2 minutes and ~48 minutes respectively. We ran all benchmarks on a Linux server (Ubuntu 12.0.4 LTS with 4 CPU, 8 GB RAM).
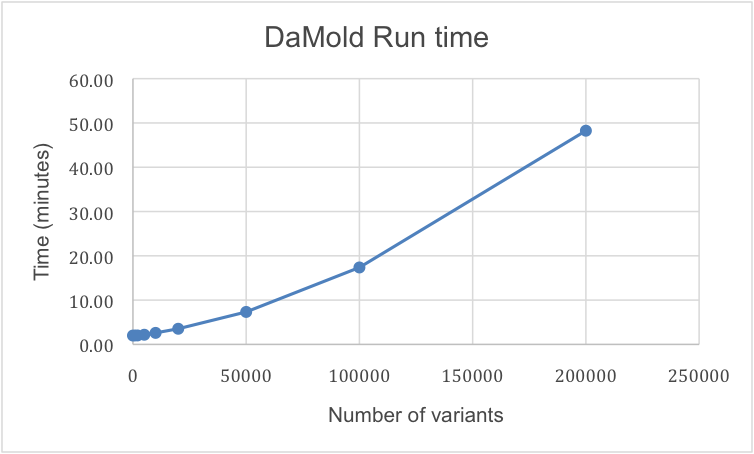
Figure 4 – Evaluation of DaMold execution time with increase of variants
Execution times were measured for different numbers of input variants (10; 100; 500; 1,000; 2,000; 5,000; 10,000; 20,000; 50,000; 100,000 and 200,000) in VCF file format. For all variants every steps of the DaMold workflow was performed (variant filtering, variant effect prediction, variant cross-referencing, and output generation). The execution time was measured in minutes. It shows that 10-500 variants can be annotated in only 2 minutes whereas in ~48 minutes 200,000 variants can be automatically annotated with DaMold.
Ram Vinay Pandey
ramvinay.pandey@gmail.com