ClinQC Wiki
ClinQC: A tool for quality control of Sanger and NGS data in clinic
Brought to you by:
ramvinay1
The user manual of ClinQC can be downloaded in PDF format from here...
ClinQC is an integrated pipeline for quality control, filtering and trimming of Sanger and NGS sequencing data for hundred to thousands of samples/patients in a single run in clinical research. It can analyze Sanger sequencing and NGS data from raw reads and produces unified output as FASTQ files per sample/patient with Sanger quality encoding. The important features of ClinQC are described below:
ClinQC supports three major NGS platforms including Illumina, 454/Roche and Ion torrent.
ClinQC supports Sanger sequencing data analysis from trace file to FASTQ file with Sanger quality encoding.
ClinQC supports Single-end and Paired-end reads.
ClinQC can be used to analyze the sequencing data generated from single-gene-panel, multigene-panel, exome-seq, genome-seq and RNA-seq experiments.
ClinQC has uniform input and output model for Sanger and NGS data analysis.
ClinQC can be used to analyze several patients/samples in a single run simultaneously.
ClinQC can be used to analyze Sanger and NGS sequencing data simultaneously in a single run.
Linux
Mac OSX 10.6 or later
Windows PC
Python 2.7.9
Biopython 1.60 or higher
Bioperl 1.6 or higher
Perl 5.10 or higher
Java 1.7 or higher
AlienTrimmer [ftp://ftp.pasteur.fr/pub/gensoft/projects/AlienTrimmer/]
TraceTuner [https://sourceforge.net/projects/tracetuner/]
FASTQC [http://www.bioinformatics.babraham.ac.uk/projects/fastqc/]
PRINSEQ [http://sourceforge.net/projects/prinseq/files/standalone/]
We provide source code in two version of ClinQC for Linux, and Macintosh operating systems. For Windows PC users we provide a fully configured Virtual Machine (VM can be used on any operating system). Along with source code and virtual machine we provide test data and extensive user manual for step-by-step get and run ClinQC for expert and non-expert users.
For all users we provide a fully configured Virtual Machine (VM), which is readily available and thus does not require any installation and configuration and works on any operating system including Windows, Linux and Mac osx. The VM can be obtained from https://sourceforge.net/p/clinqc/wiki/Virtual_Machine/
Step1.1: Download and install the Virtual Box from http://www.oracle.com/technetwork/server-storage/virtualbox/downloads/index.html#vbox
Step1.2: After installation of Virtual Box download and install the Virtual Box Extension Pack from http://www.oracle.com/technetwork/server-storage/virtualbox/downloads/index.html#extpack
Step2: Download ClinQC Virtual machine from https://sourceforge.net/p/clinqc/wiki/Virtual_Machine
Step3: Import ClinQC Virtual Machine file into Virtual Box.
Step4: Login into ClinQC Virtual machine with username and password = testuser
Step5: Open terminal and change to ClinQC_v1.0 directory with following command:
cd ~/ClinQC_v1.0
Step6: Run ClinQC pipeline with following command
sh ./run_clinqc.sh
Step7: To run ClinQC pipeline with other data analysis, prepare the Target file and ClinQCOption file to run the ClinQC pipeline.
If user wants to use ClinQC on own system then user can download the latest version of ClinQC source code (1) for Linux computers from https://sourceforge.net/projects/clinqc/files/ClinQC_v1.0-linux.zip and 2) for Macintosh OSX computers form https://sourceforge.net/projects/clinqc/files/ClinQC_v1.0-macos.zip. Move the file to an appropriate directory and run the following command to uncompress the file:
unzip ClinQC_v1.0-linux.zip
For Macintosh version run following command to unzip ClinQC source code
unzip ClinQC_v1.0-macos.zip
Note that after uncompressing the .zip file, a new folder will be created named ClinQC_v1.0. This directory contains the following files and folders. Files are denoted in blue and sub folders are denoted in red colors:

To validate the installation of the ClinQC pipeline, it can be run with a small test data set. The test data set and the corresponding ClinQC configuration files for Sanger and NGS can be obtained from https://sourceforge.net/projects/clinqc/files/test_data.zip and download in ClinQC_v1.0 folder/directory and run the following command to uncompress the file:
*unzip test_data.zip*
Note that after uncompressing the .zip file, a new folder will be created named test_data in ClinQC_v1.0 folder. And then run following two commands to run Sanger data analysis and NGS data analysis.
Sanger analysis:
cd ~/ClinQC_v1.0 ./clinqc --option_file ClinQCOptions_Sanger
NGS analysis:
cd ~/ClinQC_v1.0 ./clinqc --option_file ClinQCOptions_NGS
To get help on how to run ClinQC and required parameters enter:
./clinqc –help
ClinQC pipeline has several sequential steps that can be run by a single command. All input parameters can be specified in the ClinQCOptions file. These parameters are then used to run the whole pipeline. ClinQC provides two different input options file for Sanger and NGS:
ClinQCOptions_Sanger:* This configuration file can be used for Sanger sequencing data analysis for quality control, trimming and filtering to obtain high quality FASTQ files. We have already given default value for required parameters to run the whole Sanger sequencing analysis from raw reads to high FASTQ file.
ClinQCOptions_NGS: This configuration file can be used for NGS sequencing data analysis for quality control, trimming and filtering to obtain high quality FASTQ files. We have already given default value for required parameters to run the whole Sanger sequencing analysis from raw reads to high FASTQ file.
ClinQC can be run with the following command, which should be run under the folder/directory ClinQC_v1.0 directory:
./clinqc --option-file <path to ClinQCOptions file>
However, before running ClinQC with your own dataset all parameters have to be specified in the appropriate ClinQC Options files (ClinQCOptions_Sanger OR ClinQCOptions_NGS).
To use ClinQC for quality control, trimming and filtering Sanger sequencing files should be in AB1 and SCF format, Illumina reads in FASTQ (both paired end and single end), 454 reads in SFF and FASTA-QUAL and Ion torrent reads in SFF and FASTQ format.
Step1: Prepare Input files:
1. Prepare Target file
For each analysis user need to prepare a target file in a predefined format. It is a tab-separated text file, which contains 10 columns. As shown in figure 1, one row for each sequencing file in target file. Target file is a mandatory input, which must be provided. The target file can be given in the ClinQCOptions file with the input name Target_File=”sanger_target_file.txt”
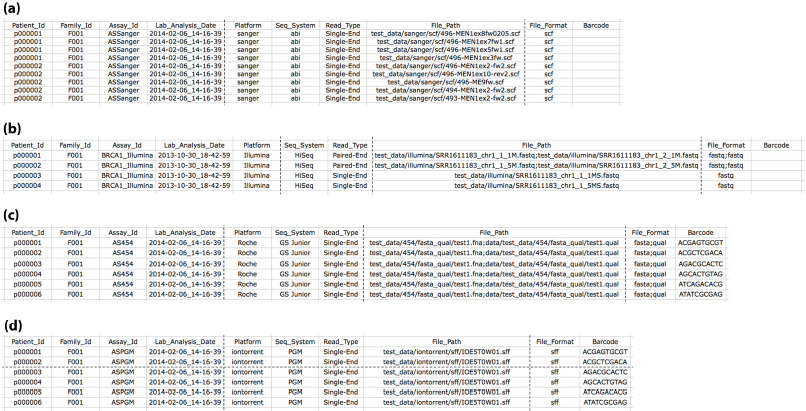
Figure 1: Shows Target files (a) Sanger target file, (2) Illumina target file, (3) 454 target file, (4) Ion torrent target
2. Prepare ClinQCOptions_Sanger file
ClinQC requires an input configuration file, which can be prepared once by customizing parameters as per the requirement, and the whole pipeline will be run without further user interaction. The ClinQC Option file is shown below:
############################################################################################################## ## ClinQC Options file (ClinQCOptions.txt) ## ## Note: This configuration file is used when user has expression data for parent1, parent2 and F1. ## ## This file sets the customizable options for running ClinQC. The options include the paths to the ## database cross reference files, paths to executables, annotation files and other parameters of the pipeline ## ## This file contains example parameter settings, which can be used to run the test data set ## provided with ClinQC. ## For running ClinQC with your own data, the example parameter values have to be modified. ## Please note that only the input value but not the parameter name itself should be changed. ## ## Note: 1) Lines starting with # are comment lines. They do not need to be changed. ## 2) Parameter's name is case sensitive. The parameters can be changed ## by replacing the values after the “=” sign. ## ############################################################################################################## # Output directory: the name of the directory can be changed. #OUTPUT_DIRECTORY = /ClinQC_1.0/sanger_output OUTPUT_DIRECTORY = test_output/sanger_test # Give the number of processors that should be used in ClinQC pipeline. THREAD = 10 ### Sequencing data quality and trimming parameters Minimum_Base_Quality = 20 Minimum_Read_Length = 50 Maximum_Read_Length = 1000 ### Sanger data quality control and trimming parameters for TTUNER Sanger_Trim_Window_Size = 10 Sanger_Trim_Base_Quality = 20 ################################################################################################################################## # # Provide the experiment information files # ################################################################################################################################## ### Target_File contains Sequencing information. (Mandetory) Target_File = test_data/sanger/scf/test_target_file.txt #Target_File = test_data/sanger/ab1/test_target_file.txt ### Provide the Primer and adapter sequences for trimming #Primer_Adapter_File = test_data/sanger/ab1/adapter_primer_sanger.txt ############################################################################################################## # # Third party software/tool executables # Provide the full path to the third party executables to run the ClinQC pipeline. # # ############################################################################################################## ###### FASTQC executables #FASTQC = ClinQC_1.0/executables/FastQC/fastqc FASTQC = executables/FastQC/fastqc ###### Sanger base calling tool #TTUNER = ClinQC_1.0/executables/tracetuner_3.0.6beta/rel/Linux/ttuner TTUNER = executables/tracetuner_3.0.6beta/rel/Linux_64/ttuner ##### AlienTrimmer is used to trim the additional sequences (Adapters and Primers) and Homopolymers #AlienTrimmer = ClinQC_1.0/executables/AlienTrimmer_0.4.0/src/AlienTrimmer.jar AlienTrimmer = executables/AlienTrimmer_0.4.0/src/AlienTrimmer.jar
Step2: Run ClinQC pipeline:
After preparing the Target File and ClinQCOptions file have been prepared and customized then ClinQC pipeline can be run with following command line
./clinqc –option_file ClinQCOptions_Sanger
Step1: Prepare Input files:
1. Prepare Target file
For each analysis user need to prepare a target file in a predefined format. It is a tab-separated text file, which contains 10 columns. As shown in below figure 5, one row for each sequencing file in target file. Target file is a mandatory input, which must be provided. The target file can be given in the ClinQCOptions file with the input name Target_File=”ngs_target_file.txt”
2. Prepare ClinQCOptions_NGS file
ClinQC requires an input configuration file, which can be prepared once by customizing parameters as per the requirement, and the whole pipeline will be run without further user interaction.
############################################################################################################## ## ClinQC Options file (ClinQCOptions.txt) ## ## Note: This configuration file is used when user has expression data for parent1, parent2 and F1. ## ## This file sets the customizable options for running ClinQC. The options include the paths to the ## database cross reference files, paths to executables, annotation files and other parameters of the pipeline ## ## This file contains example parameter settings, which can be used to run the test data set ## provided with ClinQC. ## For running ClinQC with your own data, the example parameter values have to be modified. ## Please note that only the input value but not the parameter name itself should be changed. ## ## Note: 1) Lines starting with # are comment lines. They do not need to be changed. ## 2) Parameter's name is case sensitive. The parameters can be changed ## by replacing the values after the “=” sign. ## ############################################################################################################## # Output directory: the name of the directory can be changed. #OUTPUT_DIRECTORY = /ClinQC_1.0/sanger_output OUTPUT_DIRECTORY = test_output/ngs_test # Give the number of processors that should be used in ClinQC pipeline. THREAD = 10 ### Sequencing data quality and trimming parameters Minimum_Base_Quality = 20 Minimum_Read_Length = 50 Maximum_Read_Length = 1000 ################################################################################################################################## # # Provide the experiment information files # ################################################################################################################################## ### Target_File contains Sequencing information. (optional) #Target_File = test_data/iontorrent/sff/test_target_file.txt Target_File = test_data/illumina/Illumina_target_file.txt ### Provide the Primer and adapter sequences for trimming #Primer_Adapter_File = test_data/iontorrent/sff/adapter_primer.txt ############################################################################################################## # # Third party software/tool executables # Provide the full path to the third party executables to run the ClinQC pipeline. # # ############################################################################################################## ###### FASTQC executables #FASTQC = ClinQC_1.0/executables/FastQC/fastqc FASTQC = executables/FastQC/fastqc ##### AlienTrimmer is used to trim the additional sequences (Adapters and Primers) and Homopolymers #AlienTrimmer = ClinQC_1.0/executables/AlienTrimmer_0.4.0/src/AlienTrimmer.jar AlienTrimmer = executables/AlienTrimmer_0.4.0/src/AlienTrimmer.jar ##### PRINSEQ to filter PCR duplicates and contamination filtering. PRINSEQ = executables/prinseq-lite-0.20.4/prinseq-lite.pl
Step2: Run ClinQC pipeline:
After preparing the Target File and ClinQCOptions file have been prepared and customized then ClinQC pipeline can be run with following command line
./clinqc –option_file ClinQCOptions_NGS
After running ClinQC, the results of the ClinQC pipeline can be found in the output directory (as specified in the ClinQC Options file). Results are provided in the following format.
<OUTPUT_FILE>
<OUTPUT_DIR>
|
- <fastq_files>
|
- <patient1_1.fq;patient1_2.fq>
- <patient2_1.fq;patient2_2.fq>
- <patient3_1.fq;patient3_2.fq>
- .
- .
- .
- <patientN_1.fq;patientN_2.fq>
- <QC_report>
|
- <before_qc_patient1_1.fq_fastqc_qc_report.html>
- <after_qc_patient1_1.fq_fastqc_qc_report.html>
- <before_qc_patient1_2.fq_fastqc_qc_report.html>
- <after_qc_patient1_2.fq_fastqc_qc_report.html>
- .
- .
- |
- .
- <before_qc_patientN_1.fq_fastqc_qc_report.html>
- <after_qc_patientN_1.fq_fastqc_qc_report.html>
- <before_qc_patientN_2.fq_fastqc_qc_report.html>
- <after_qc_patientN_2.fq_fastqc_qc_report.html>
<fastq_files>:
This output folder contains high quality FASTQ file for each patient with Sanger quality encoding. If reads are in paired-end then there will be two files for each patient.
<QC_report>:
This output folder contains Quality control and trimming report in html format generated by FASTQC tool. There are two files for each FASTQ files 1) before quality control and 2) after quality control.
Ram Vinay Pandey
ramvinay.pandey@gmail.com