In this HowTo we describe how Elk should be compiled for maximum performance on a multi-node cluster.
Elk uses both the Message Passing Interface (MPI) and OpenMP for parallelism as well as highly optimised libraries for linear algebra and the fast Fourier transform (FFT). Compilation may also include linking to the Libxc and Wannier90 libraries.
We will also show how to run the code across multiple nodes using the mpirun command and which OpenMP environment variables should be set.
Considerable effort is put into optimising the Elk and each new version is usually faster than the last. Always try to compile your own code from scratch using latest official release (found here http://sourceforge.net/projects/elk/files/) rather than using a pre-compiled package. This way the executable will be tailored for your particular computer system.
For this HowTo we will use Elk version 7.1.14. Download elk-7.1.14.tgz and unpack it.
Last edit: J. K. Dewhurst 2021-03-18
If you would like to refer to this comment somewhere else in this project, copy and paste the following link:
Compiling and running software on a cluster can be a challenge because of the different ways servers are set up.
The first thing to do is to find out which compilers are available. Type
moduleavail
Find which package corresponds to the latest MPI-compatible Fortran compiler. This will most likely be Intel Fortran or GNU Fortran. You will also need optimised libraries for BLAS and LAPACK. These can include MKL from Intel (https://software.intel.com/en-us/mkl), OpenBLAS (https://www.openblas.net/) or BLIS (https://developer.amd.com/amd-aocl/blas-library/).
We find that MKL is generally faster on Intel computers but OpenBLAS and BLIS are faster on AMD machines.
You will also require an optimised FFT library. Elk can use either FFTW version 3.x or the native FFT routines in MKL. FFTW can be used either as a standalone library or as part of MKL.
Now type
modulepurge
followed by
moduleload<package>
where <package> is the desired compiler and library.
You may have to contact the system administrator for assistance with finding or installing the correct compiler and libraries.
Check that you now have an MPI compatible compiler. The command is usually mpif90, mpifort or mpiifort. We will assume mpif90 from now on.
Type
mpif90 --version
This should write the name and version of the compiler. For example:
ifort (IFORT) 18.0.0 20170811
Copyright (C) 1985-2017 Intel Corporation. All rights reserved.
Last edit: J. K. Dewhurst 2019-09-01
If you would like to refer to this comment somewhere else in this project, copy and paste the following link:
Before we compile Elk, we should first download and compile the libraries Libxc (http://www.tddft.org/programs/libxc/) and Wannier90 (http://www.wannier.org/).
Libxc first: we're using version 5.1.2 for this HowTo. This will change in the future so check the Elk manual to find the latest compatible version.
Download and unpack libxc-5.1.2.tar.gz. In the directory libxc-5.1.2, run
./configure
If no errors are reported then run
make
Assuming that this compiles correctly, two static library files should then be copied to the Elk source directory:
Note the code generated with the -xCORE-AVX2 option will work work only on specific Intel processors. You should check which type of processors are available on your cluster. These are listed in the file /proc/cpuinfo.
Options for various Intel processor types:
-xSSE4.2 can run on Westmere, Sandy Bridge, Ivy Bridge, Haswell, Broadwell, Skylake, and Cascade Lake processors
-xAVX can run on Sandy Bridge, Ivy Bridge, Haswell, Broadwell, Skylake, and Cascade Lake processors
-xCORE-AVX2 can run on Haswell, Broadwell, Skylake, and Cascade Lake processors
-xCORE-AVX512 can run only on Skylake and Cascade Lake processors
Last edit: J. K. Dewhurst 2021-03-18
If you would like to refer to this comment somewhere else in this project, copy and paste the following link:
This compilation also uses MKL (which is freely available from Intel) for LAPACK and FFTW but uses BLIS for BLAS calls. In our experience, this is slightly faster than using MKL for BLAS.
By using MKL for LAPACK calls we can exploit its efficient multi-threaded eigenvalue solvers for large matrices. BLIS is better optimised than MKL for Zen+ and Zen2 architectures which is why we use it for BLAS calls.
Last edit: J. K. Dewhurst 2021-03-18
If you would like to refer to this comment somewhere else in this project, copy and paste the following link:
Elk is a hybrid code and runs with both OpenMP and MPI. On a multi-node cluster MPI is used between nodes and OpenMP is used for multi-threading among the CPU cores. Here is a schematic of the running code:
MPI runs several copies of the same code which then pass messages between one another in order to decide on what to calculate, whereas OpenMP spawns multiple threads when it encounters a parallel loop or section. MPI is used mainly for k-point parallelism whereas OpenMP is used for both k-points and finer grained parallelism over states, atoms, etc.
Most codes are run with a batch queuing system on a shared cluster such as Grid Engine or Slurm. You'll need to find out which is available on your computer from the system administrator.
The actual command for running Elk across multiple nodes is mpirun. Here is the one we use on our cluster:
This tells the MPI system to use Infiniband to communicate and also specifies the number of OpenMP threads allowed on each node but doesn't bind those threads to particular cores. Elk can use hundreds cores efficiently so long as there are a sufficient number of k-points.
We have also found that it is better to bind threads to certain cores in some circumstances. For example, on our 32 core AMD 2990WX Threadripper machines using
Elk uses the enviroment OMP_NUM_THREADS to decide how many OpenMP threads to use. This can be overridden by using the variable maxthd in elk.in. If maxthd is a positive number then the number of threads is set equal to that. However when
maxthd
-1
then the number of threads is equal to number of processors, although this is usually the
number of physical threads (including hyperthreads) which may not be the most efficient. Note also that solid state electronic structure codes will be often memory-bound and not compute-bound. In other words the CPU scaling is limited by how quickly the code can access memory and not how fast it can perform floating point operations. This is because each k-point requires its own set up of the Hamiltonian matrix and diagonalisation and may need hundreds of megabytes of memory. Thus computers with more memory channels per CPU will generally perform better.
Elk will ignore the environment variables OMP_NESTED, OMP_DYNAMIC and OMP_MAX_ACTIVE_LEVELS. Instead, the maximum parallel nesting level is set by the variable maxlvl (default 4) and the number of threads for each loop is determined from the number of active threads in that nesting level. See the module modomp.f90 for details. Finally, the number of threads used by MKL for certain LAPACK calls is set by the variable maxthdmkl. By default this is set to 8.
Last edit: J. K. Dewhurst 2021-04-04
If you would like to refer to this comment somewhere else in this project, copy and paste the following link:
I've experienced that if I use more than one mpi tasks for a Gamma-pt calculation, the code crashes. But works well if I use mpirun with "-n 1" or simply submit a serial run (without mpirun). Is this expected?
Also, in the point above, you've commented that the mpi parallelism is only across kpoints. In that case, will the code not run into wall time issues if we deal with slightly larger simulation cells (for example,point-vacancy systems where the simulation domain is increased to account for spurious vacancy-vacancy interaction because of periodic BCs). What is the typical max number of atoms one can simulate using elk for ground state calculations?
If you would like to refer to this comment somewhere else in this project, copy and paste the following link:
You're correct: MPI is only used for k-points or q-points (and the FFT in ultra-long range calculations).
I probably wouldn't use Elk for more than 200 atoms, depending on the type. You'd be better off using another code like VASP.
For very large unit cells, you can increase the number of threads used by MKL with
maxthdmkl
24
This will use 24 threads for the diagonalisation step. Alternatively iterative diagonalisation can be enabled for the first-variational step:
tefvit
.true.
One of the slowest parts is the setup of the APW-APW part of the Hamiltonian and unfortunately there's no easy way to parallelise this. Some early versions of Elk were able to apply the Hamiltonian to a vector (like a pseudopotential code) and run in parallel but this never worked very well so we removed it.
You can also enable low-quality calculations with
lowq
.true.
which will run faster for large systems at the expense of accuracy.
Finally, there is a new feature: ultra long-range calculations where the number of atoms is virtually unlimited (we're running a Skyrmion with 157500 atoms at the moment). There are however restrictions of the types of systems that can be run: the nuclei have to be periodic but the electronic state can have long-range modulations. This is still undergoing testing and development but is available in 6.2.8.
Regards,
Kay.
If you would like to refer to this comment somewhere else in this project, copy and paste the following link:
Despite what I said, I've mandaged to speed up the APW-APW part of the first-variational Hamiltonian an overlap matrices and also make it parallel. This is particularly apparent for large systems where you can get an order-of-magnitude speed up.
This is now available in version 6.3.2 of the code.
Kay.
If you would like to refer to this comment somewhere else in this project, copy and paste the following link:
This thread is open to anyone who would like to post their own compilation options, submission scripts and environment variables which are optimal for their computer system and also to report any problems encountered during compilation.
If you would like to refer to this comment somewhere else in this project, copy and paste the following link:
As of now, I am doing some tests regarding parallel execution of elk.
I will post the results here.
So far - I must say that some amazing work was done for 6.3.2 - it is about THREE times faster than 6.2.8 with the same compilation.
Developers, you are great!
The improvements, however, come at a price - the code shows some new kind memory sensitivity.
I.e., if the memory is almost enought, but not abundant, is works slower than 6.2.8.
Thus, if your job was using 90% and more of the allocated memory - try running it again, with 50% more memory.
Best regards.
Andrew
Last edit: Andrew Shyichuk 2019-09-29
If you would like to refer to this comment somewhere else in this project, copy and paste the following link:
Thanks Andrew, glad to hear it! It's probably something we should have done a while ago...
If you are running out of memory, you can restrict the number of threads allowed a the first nesting level with:
maxthd112
for example. The calculation will still use all available threads but only 12 of those will be at the first level (usually a k-point loop), which take the most memory.
Regards,
Kay.
If you would like to refer to this comment somewhere else in this project, copy and paste the following link:
The cluster may have this kind of default setting simply because it was overlooked, but it also might be for a reason (although it is a strange thing to have on cluster).
Thus, just to be consistent, it is better to contact the cluster admins and make sure that users may increase OMP_NUM_THREADS. There might be even some manual regarding recommend settings for the submitted jobs.
Best regards.
Andrew
If you would like to refer to this comment somewhere else in this project, copy and paste the following link:
I've made a short benchmark of different FFT routines on a Ryzen machine.
I've used YBCO and CuFeS2 inputs from Elk basic examples, with increased k-point grids for a longer run time.
Elk was compiled with GCC 9 with built-in libs, except for FFT, which was either:
- built-in
- precompiled FFTW 3.3.8, from repository (Fedora 30 Server)
- manually compiled FFTW 3.3.8 (--enable-sse2 --enable-avx2 )
Workin directroies and scratch were located on a NVMe RAID 0 (stripped volume), and nwrite was set to 0, thus reducing the effects of write lags.
The machine load with other tasks was low and not much changing during the test.
I used either 1 or 4 threads for the Elk tasks.
Total run times, user times and time per iteration were compared.
The tests were repeated 5 times each.
Detailed reaults are in the attached spreadsheet.
Briefly, with respect to the built-in FFT, the manually compiled FFTW was performing best, with top gain of 4%.
But, the respective standard deviation in the compared data was something like 3-7%.
The precompiled FFTW was sometimes better, and sometimes worse, by about 1-2%.
The 4-thread gains were larger than those from 1 thread.
The conclusion is thus: manually-compiled FFTW (with vectorization) is faster than the built-in one, but the difference is not drastic, and comparable to the "experimental noise".
Note that the latest versions of Elk (6.8.4 and above) use stack space more aggressively. This is because accessing variables on the stack is generally faster than on the heap. The reasons for this are a) lower overhead by avoiding allocate and deallocate statements, which require thread locking b) the less fragmented page layout of the stack and c) increased likelihood that the stack will be in the processors cache at any given time.
This increased reliance on the stack may cause the code to crash (usually with a segmentation fault) if there is not enough stack space for a thread. It is therefore important to increase the size of the stack space per thread by setting the environment variable OMP_STACKSIZE. A value of between 50 and 100 MB should suffice for most tasks.
This can be done with the command
exportOMP_STACKSIZE=64M
which will affect subsequent Elk runs.
Multi-node machines require that this environment variable be passed to all processes. On our cluster we use the OpenMPI implementation of the command
I'm having problems compiling elk-7.2.42 on our cluster. I'm using gnu-4.8.1 compilers.
The error occurs with splined.f90 and the output is
"splined.f90:18.6:
!$OMP SIMD LASTPRIVATE(f1,f2,f3,f4)
1
Error: OpenMP directives at (1) may not appear in PURE or ELEMENTAL procedures"
I don't recall seeing this error when compiling older versions of elk on the same cluster.
My make.inc is essentially the same as in the example for AMD processors and gnu compilers posted above on September 2019. The only difference is that I don't use the compiler option "-ffpe-summary=none" for f90 or f77 as it is unknown on our system.
Any ideas?
Best regards,
Kari
If you would like to refer to this comment somewhere else in this project, copy and paste the following link:
In this HowTo we describe how Elk should be compiled for maximum performance on a multi-node cluster.
Elk uses both the Message Passing Interface (MPI) and OpenMP for parallelism as well as highly optimised libraries for linear algebra and the fast Fourier transform (FFT). Compilation may also include linking to the Libxc and Wannier90 libraries.
We will also show how to run the code across multiple nodes using the mpirun command and which OpenMP environment variables should be set.
Considerable effort is put into optimising the Elk and each new version is usually faster than the last. Always try to compile your own code from scratch using latest official release (found here http://sourceforge.net/projects/elk/files/) rather than using a pre-compiled package. This way the executable will be tailored for your particular computer system.
For this HowTo we will use Elk version 7.1.14. Download elk-7.1.14.tgz and unpack it.
Last edit: J. K. Dewhurst 2021-03-18
Compiling Elk
Compiling and running software on a cluster can be a challenge because of the different ways servers are set up.
The first thing to do is to find out which compilers are available. Type
Find which package corresponds to the latest MPI-compatible Fortran compiler. This will most likely be Intel Fortran or GNU Fortran. You will also need optimised libraries for BLAS and LAPACK. These can include MKL from Intel (https://software.intel.com/en-us/mkl), OpenBLAS (https://www.openblas.net/) or BLIS (https://developer.amd.com/amd-aocl/blas-library/).
We find that MKL is generally faster on Intel computers but OpenBLAS and BLIS are faster on AMD machines.
You will also require an optimised FFT library. Elk can use either FFTW version 3.x or the native FFT routines in MKL. FFTW can be used either as a standalone library or as part of MKL.
Now type
followed by
where
<package>is the desired compiler and library.You may have to contact the system administrator for assistance with finding or installing the correct compiler and libraries.
Check that you now have an MPI compatible compiler. The command is usually mpif90, mpifort or mpiifort. We will assume mpif90 from now on.
Type
This should write the name and version of the compiler. For example:
Last edit: J. K. Dewhurst 2019-09-01
Before we compile Elk, we should first download and compile the libraries Libxc (http://www.tddft.org/programs/libxc/) and Wannier90 (http://www.wannier.org/).
Libxc first: we're using version 5.1.2 for this HowTo. This will change in the future so check the Elk manual to find the latest compatible version.
Download and unpack libxc-5.1.2.tar.gz. In the directory libxc-5.1.2, run
./configureIf no errors are reported then run
makeAssuming that this compiles correctly, two static library files should then be copied to the Elk source directory:
cp src/.libs/libxc.a src/.libs/libxcf90.a ~/elk-7.1.14/src/Next is Wannier90: download and unpack wannier90-3.1.0.tar.gz (or the latest compatible version).
In the directory wannier90-3.1.0 copy the appropriate make.inc from the config directory. Here we'll use the Intel version:
cp config/make.inc.ifort ./make.incThen run
makeFollowed by
make libCopy the library file to the Elk source directory:
cp libwannier.a ~/elk-7.1.14/src/Last edit: J. K. Dewhurst 2021-03-18
Now to compile Elk. On our Intel cluster, we use Intel Fortran with the following make.inc file:
This uses MKL for BLAS, LAPACK and FFTW.
You will also need to copy the file mkl_dfti.f90 from the MKL include directory to the Elk source directory. For example
cp /usr/include/mkl/mkl_dfti.f90 ~/elk-7.1.14/src/Note the code generated with the -xCORE-AVX2 option will work work only on specific Intel processors. You should check which type of processors are available on your cluster. These are listed in the file /proc/cpuinfo.
Options for various Intel processor types:
-xSSE4.2 can run on Westmere, Sandy Bridge, Ivy Bridge, Haswell, Broadwell, Skylake, and Cascade Lake processors
-xAVX can run on Sandy Bridge, Ivy Bridge, Haswell, Broadwell, Skylake, and Cascade Lake processors
-xCORE-AVX2 can run on Haswell, Broadwell, Skylake, and Cascade Lake processors
-xCORE-AVX512 can run only on Skylake and Cascade Lake processors
Last edit: J. K. Dewhurst 2021-03-18
On our AMD machines we use GNU Fortran with the following make.inc file:
This compilation also uses MKL (which is freely available from Intel) for LAPACK and FFTW but uses BLIS for BLAS calls. In our experience, this is slightly faster than using MKL for BLAS.
By using MKL for LAPACK calls we can exploit its efficient multi-threaded eigenvalue solvers for large matrices. BLIS is better optimised than MKL for Zen+ and Zen2 architectures which is why we use it for BLAS calls.
Last edit: J. K. Dewhurst 2021-03-18
Running Elk
Elk is a hybrid code and runs with both OpenMP and MPI. On a multi-node cluster MPI is used between nodes and OpenMP is used for multi-threading among the CPU cores. Here is a schematic of the running code:
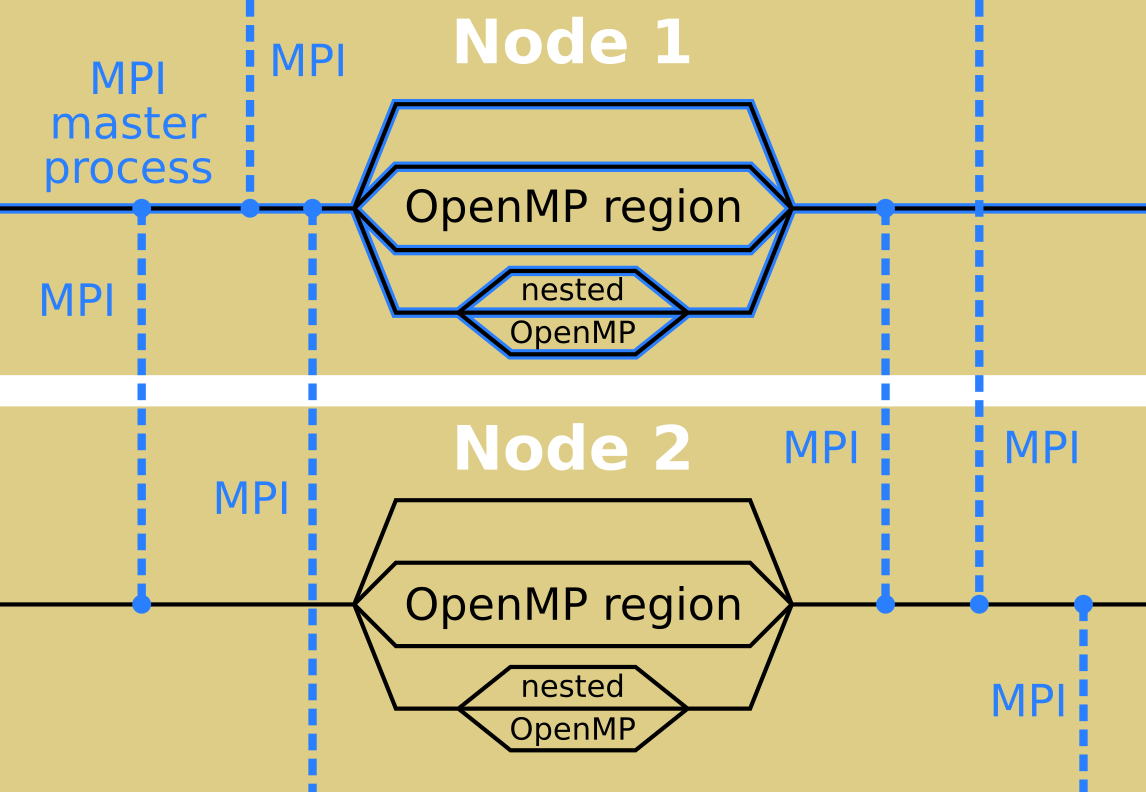
MPI runs several copies of the same code which then pass messages between one another in order to decide on what to calculate, whereas OpenMP spawns multiple threads when it encounters a parallel loop or section. MPI is used mainly for k-point parallelism whereas OpenMP is used for both k-points and finer grained parallelism over states, atoms, etc.
Most codes are run with a batch queuing system on a shared cluster such as Grid Engine or Slurm. You'll need to find out which is available on your computer from the system administrator.
The actual command for running Elk across multiple nodes is mpirun. Here is the one we use on our cluster:
This tells the MPI system to use Infiniband to communicate and also specifies the number of OpenMP threads allowed on each node but doesn't bind those threads to particular cores. Elk can use hundreds cores efficiently so long as there are a sufficient number of k-points.
We have also found that it is better to bind threads to certain cores in some circumstances. For example, on our 32 core AMD 2990WX Threadripper machines using
gives a small speedup.
Elk uses the enviroment OMP_NUM_THREADS to decide how many OpenMP threads to use. This can be overridden by using the variable maxthd in elk.in. If maxthd is a positive number then the number of threads is set equal to that. However when
then the number of threads is equal to number of processors, although this is usually the
number of physical threads (including hyperthreads) which may not be the most efficient. Note also that solid state electronic structure codes will be often memory-bound and not compute-bound. In other words the CPU scaling is limited by how quickly the code can access memory and not how fast it can perform floating point operations. This is because each k-point requires its own set up of the Hamiltonian matrix and diagonalisation and may need hundreds of megabytes of memory. Thus computers with more memory channels per CPU will generally perform better.
Elk will ignore the environment variables OMP_NESTED, OMP_DYNAMIC and OMP_MAX_ACTIVE_LEVELS. Instead, the maximum parallel nesting level is set by the variable maxlvl (default 4) and the number of threads for each loop is determined from the number of active threads in that nesting level. See the module modomp.f90 for details. Finally, the number of threads used by MKL for certain LAPACK calls is set by the variable maxthdmkl. By default this is set to 8.
Last edit: J. K. Dewhurst 2021-04-04
I've experienced that if I use more than one mpi tasks for a Gamma-pt calculation, the code crashes. But works well if I use mpirun with "-n 1" or simply submit a serial run (without mpirun). Is this expected?
Also, in the point above, you've commented that the mpi parallelism is only across kpoints. In that case, will the code not run into wall time issues if we deal with slightly larger simulation cells (for example,point-vacancy systems where the simulation domain is increased to account for spurious vacancy-vacancy interaction because of periodic BCs). What is the typical max number of atoms one can simulate using elk for ground state calculations?
I'll take a look at the problem - thanks.
You're correct: MPI is only used for k-points or q-points (and the FFT in ultra-long range calculations).
I probably wouldn't use Elk for more than 200 atoms, depending on the type. You'd be better off using another code like VASP.
For very large unit cells, you can increase the number of threads used by MKL with
This will use 24 threads for the diagonalisation step. Alternatively iterative diagonalisation can be enabled for the first-variational step:
One of the slowest parts is the setup of the APW-APW part of the Hamiltonian and unfortunately there's no easy way to parallelise this. Some early versions of Elk were able to apply the Hamiltonian to a vector (like a pseudopotential code) and run in parallel but this never worked very well so we removed it.
You can also enable low-quality calculations with
which will run faster for large systems at the expense of accuracy.
Finally, there is a new feature: ultra long-range calculations where the number of atoms is virtually unlimited (we're running a Skyrmion with 157500 atoms at the moment). There are however restrictions of the types of systems that can be run: the nuclei have to be periodic but the electronic state can have long-range modulations. This is still undergoing testing and development but is available in 6.2.8.
Regards,
Kay.
Thanks for clearing that up
Hi again,
Despite what I said, I've mandaged to speed up the APW-APW part of the first-variational Hamiltonian an overlap matrices and also make it parallel. This is particularly apparent for large systems where you can get an order-of-magnitude speed up.
This is now available in version 6.3.2 of the code.
Kay.
Thanks for the information. I'll try it out.
This thread is open to anyone who would like to post their own compilation options, submission scripts and environment variables which are optimal for their computer system and also to report any problems encountered during compilation.
Dear Users,
As of now, I am doing some tests regarding parallel execution of elk.
I will post the results here.
So far - I must say that some amazing work was done for 6.3.2 - it is about THREE times faster than 6.2.8 with the same compilation.
Developers, you are great!
The improvements, however, come at a price - the code shows some new kind memory sensitivity.
I.e., if the memory is almost enought, but not abundant, is works slower than 6.2.8.
Thus, if your job was using 90% and more of the allocated memory - try running it again, with 50% more memory.
Best regards.
Andrew
Last edit: Andrew Shyichuk 2019-09-29
Thanks Andrew, glad to hear it! It's probably something we should have done a while ago...
If you are running out of memory, you can restrict the number of threads allowed a the first nesting level with:
for example. The calculation will still use all available threads but only 12 of those will be at the first level (usually a k-point loop), which take the most memory.
Regards,
Kay.
Dear Kay,
As I understand, maxthd1 will limit any first-level parallelization, the k-point loop or not.
Is there maybe a keyword to specifically limit k-point parallelization only?
I am trying to run RDMFT calculation and I get the impression that maxthd1 also limits density matrix (KMAT.OUT) calculation.
Best regards.
Andrew
Dear Kay,
If on our cluster the enviroment OMP_NUM_THREADS=1, does this mean the OpenMP can not be used?
Best,
Jin
Dear Kay,
If on our cluster the enviroment OMP_NUM_THREADS=1, does this mean the OpenMP can not be used?
Best,
Jin
Hi Jin,
You can set the enviroment yourself with
(or however many cores you have)
Alternatively, you can set the number of threads in elk.in with
Elk can also set the number of threads equal to the number of cores with
Regards,
Kay.
Dear Jin,
Dear Kay,
I must add a small coment.
The cluster may have this kind of default setting simply because it was overlooked, but it also might be for a reason (although it is a strange thing to have on cluster).
Thus, just to be consistent, it is better to contact the cluster admins and make sure that users may increase OMP_NUM_THREADS. There might be even some manual regarding recommend settings for the submitted jobs.
Best regards.
Andrew
Good point: check with the sysadmin first to see if it's OK to use more than one thread per node.
You can still run Elk in MPI mode only but it is not as efficient because MPI is used almost exclusively for k-points.
Regards,
Kay.
Dear Users,
I've made a short benchmark of different FFT routines on a Ryzen machine.
I've used YBCO and CuFeS2 inputs from Elk basic examples, with increased k-point grids for a longer run time.
Elk was compiled with GCC 9 with built-in libs, except for FFT, which was either:
- built-in
- precompiled FFTW 3.3.8, from repository (Fedora 30 Server)
- manually compiled FFTW 3.3.8 (--enable-sse2 --enable-avx2 )
Workin directroies and scratch were located on a NVMe RAID 0 (stripped volume), and nwrite was set to 0, thus reducing the effects of write lags.
The machine load with other tasks was low and not much changing during the test.
I used either 1 or 4 threads for the Elk tasks.
Total run times, user times and time per iteration were compared.
The tests were repeated 5 times each.
Detailed reaults are in the attached spreadsheet.
Briefly, with respect to the built-in FFT, the manually compiled FFTW was performing best, with top gain of 4%.
But, the respective standard deviation in the compared data was something like 3-7%.
The precompiled FFTW was sometimes better, and sometimes worse, by about 1-2%.
The 4-thread gains were larger than those from 1 thread.
The conclusion is thus: manually-compiled FFTW (with vectorization) is faster than the built-in one, but the difference is not drastic, and comparable to the "experimental noise".
Note that the latest versions of Elk (6.8.4 and above) use stack space more aggressively. This is because accessing variables on the stack is generally faster than on the heap. The reasons for this are a) lower overhead by avoiding allocate and deallocate statements, which require thread locking b) the less fragmented page layout of the stack and c) increased likelihood that the stack will be in the processors cache at any given time.
This increased reliance on the stack may cause the code to crash (usually with a segmentation fault) if there is not enough stack space for a thread. It is therefore important to increase the size of the stack space per thread by setting the environment variable OMP_STACKSIZE. A value of between 50 and 100 MB should suffice for most tasks.
This can be done with the command
which will affect subsequent Elk runs.
Multi-node machines require that this environment variable be passed to all processes. On our cluster we use the OpenMPI implementation of the command
to achieve this. The convention for passing enviroment variables, however, differs between implementations.
Hello,
I'm having problems compiling elk-7.2.42 on our cluster. I'm using gnu-4.8.1 compilers.
The error occurs with splined.f90 and the output is
"splined.f90:18.6:
!$OMP SIMD LASTPRIVATE(f1,f2,f3,f4)
1
Error: OpenMP directives at (1) may not appear in PURE or ELEMENTAL procedures"
I don't recall seeing this error when compiling older versions of elk on the same cluster.
My make.inc is essentially the same as in the example for AMD processors and gnu compilers posted above on September 2019. The only difference is that I don't use the compiler option "-ffpe-summary=none" for f90 or f77 as it is unknown on our system.
Any ideas?
Best regards,
Kari
Please help me how to compile this code. I'm unable to compile it. Its showing error after running make all command as shown in the figure.